Årtiers forskning sat under pres af en ny hypotese
I årtier har videnskaben fokuseret på at fjerne beta-amyloid-plaques fra hjernen. Men hvad nu hvis det egentlige problem befinder sig et helt andet sted – inde i neuronerne selv, hvor beta-amyloid og proteinet tau kæmper om kontrollen over cellernes transportsystemer?
Nye studier fra University of California Riverside antyder, at sygdommens årsag måske ikke ligger i selve proteinaflejringerne, men i en intens konkurrence, der udspiller sig dybt inde i nervecellerne. I centrum for denne hypotese står to velkendte “mistænkte”: beta-amyloid og proteinet tau.
Hvorfor har jagten på beta-amyloid-plaques ikke givet de ventede resultater?
Den klassiske forklaring på Alzheimers sygdom var enkel. Beta-amyloid-plaques og klumper af proteinet tau ophobes i hjernen, beskadiger neuronerne og forårsager dermed demens. Logikken virkede indlysende – når noget sætter sig fast, skal det fjernes. Derfor afprøvede hundredvis af eksperimentelle behandlinger netop denne strategi.
Resultatet var imidlertid nedslående. På trods af milliarder af dollars investeret i forskning lykkedes det i bedste fald at bremse sygdomsforløbet en smule – og ofte slet ikke. Farmaceutiske selskaber udviklede antistoffer, der kunne binde beta-amyloid og fjerne det fra hjernevævet. Nogle præparater reducerede faktisk mængden af plaques markant. Alligevel viste patienterne sjældent nogen væsentlig forbedring af de kognitive funktioner. Hos en del patienter opstod der endda bivirkninger som betændelse og blødninger i hjernen.
Et vigtigt observationsfund har desuden forvirret forskerne: Nogle ældre mennesker har betydelige mængder beta-amyloid-plaques i hjernen, men udviser ingen tegn på demens. Til gengæld korrelerer sygdommens sværhedsgrad bedst med tilstedeværelsen af det patologiske tau-protein inde i neuronerne.
Beta-amyloid mod tau: en kamp om mikrotubuli inde i neuronen
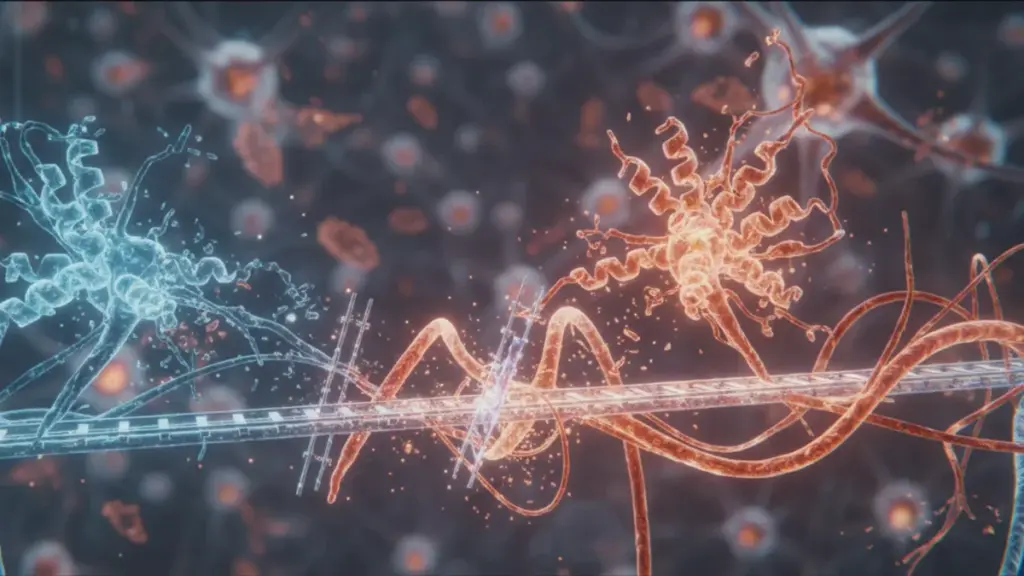
Teamet under ledelse af Ryan Julian fra University of California Riverside rettede derfor opmærksomheden mod, hvad der foregår inde i nervecellerne. Deres hypotese bygger på den antagelse, at beta-amyloid og tau konkurrerer om de samme bindingssteder på mikrotubuli – strukturer der er afgørende for neuronernes overlevelse.
Mikrotubuli er tynde rørlignende strukturer, der fungerer som cellens transportsystem. Ad disse “motorveje” bevæger proteiner, vesikler med neurotransmittere og andet vigtigt gods sig rundt. Uden et velfungerende transportsystem begynder nervecellen langsomt at kvæles og dø.
Tau-proteinets normale funktion er at stabilisere disse mikrotubuli – forestil dig det som specialbeslag og klemmer, der holder rørene på plads og i god stand. Når tau fungerer korrekt, kører hjernens kommunikationssystem gnidningsløst. Neuronerne kan sende signaler, forsyne sig med energi og fjerne affaldsstoffer effektivt.
Ryan Julians team undersøgte nøje de steder, hvor tau hæfter sig til mikrotubuli. Det viste sig, at de tau-fragmenter, der er ansvarlige for denne binding, er overraskende lig de sekvenser, der findes i beta-amyloid – både hvad angår størrelse og struktur. Det fik forskerne til at formode, at begge proteiner kan konkurrere om de samme bindingssteder.
For at forstå konsekvenserne kan man forestille sig mikrotubuli som skinnerne i en neuron. Tau er svellerne og befæstningselementerne, der sikrer banens stabilitet. Beta-amyloid optræder i denne metafor som en sabotør, der forsøger at besætte de pladser, der er beregnet til tau. Lykkes det, begynder trafikken at havarere.
Fluorescenstest afslørede overraskende egenskaber ved beta-amyloid
For at verificere, hvad strukturligheden betyder i praksis, mærkede forskerne beta-amyloid og tau med fluorescerende markører og fulgte deres adfærd under laboratorieforhold. Resultatet var entydigt: beta-amyloid hæfter sig også til mikrotubuli – og gør det med en styrke, der kan sammenlignes med tau’s.
Når der er for meget beta-amyloid til stede, begynder det at fortrænge tau fra mikrotubuli. Neuronerne mister derved deres stabile “transportskelet”, og den interne molekylære transport bliver forstyrret. Transporten af neurotransmittere som dopamin, serotonin og acetylcholin aftager eller stopper helt.
Set fra dette perspektiv handler sygdommen ikke blot om ophobning af aflejringer, men om en forstyrret magtbalance mellem to proteiner, der kæmper om de samme bindingssteder. De ydre beta-amyloid-plaques, der er synlige på hjernebilleder, kan dermed snarere være et udtryk for et generelt proteinkaoset end selve dræberen af cellerne.
Det afgørende slag leveres af den indre konkurrence om mikrotubuli. Når beta-amyloid trænger ind i neuronen og overtager kontrollen, “løber tau af sporet”, begynder at danne aggregater og bevæger sig til steder, hvor det virker destruktivt. Dette scenarie forklarer bedre, hvorfor nogle mennesker med plaques i hjernen forbliver kognitivt raske – deres beta-amyloid forbliver uden for neuronerne.
Den nye model forklarer tidligere modstridende data om Alzheimers sygdom
Det californiske team tilbyder en forklaring på flere tidligere modstridende observationer. På den ene side ved vi, at en del personer har beta-amyloid-plaques i hjernen, men aldrig udvikler fuldt fremskreden Alzheimer. På den anden side korrelerer tilstedeværelsen af patologisk tau stærkt med symptomernes intensitet.
Forskerne foreslår følgende forklaring: de plaques, der er synlige på hjernebilleder, dannes primært uden for neuronerne. Det egentlige drama udspiller sig imidlertid inde i cellerne. Når beta-amyloid trænger ind i en neuron, begynder det at konkurrere med tau om mikrotubuli. Den interne transport bliver kaotisk, og tau “løber af sporet”.
I dette scenarie er de ydre plaques snarere et symptom på et generelt proteinkaos i hjernen end en direkte cellekiller. Denne betragtning hjælper med at forklare, hvorfor fjernelse af plaques ofte ikke medfører dramatisk bedring – den egentlige kamp finder sted et andet sted.
En anden vigtig faktor er autofagi – cellernes naturlige oprydningssystem, der nedbryder beskadigede proteiner. Hos et ungt, sundt menneske nedbryder og fjerner denne mekanisme effektivt overskydende beta-amyloid. Med alderen mister autofagien sin ydeevne. Beskadigede proteiner cirkulerer længere, og beta-amyloid begynder hurtigere at ophobes inde i neuronerne.
Jo mere der er inde i cellen, jo større er presset på mikrotubuli og fortrængningen af tau. Denne kæde af begivenheder forklarer godt, hvorfor alder er den stærkeste risikofaktor for Alzheimers sygdom, og hvorfor sygdommen så ofte hænger sammen med en ophobning af mange små skader frem for et enkelt “slag”.
Lithium som spor: måske skal man beskytte transportvejene frem for at fjerne trafikpropper
Et interessant bidrag til diskussionen om mikrotubuli er studier af lithium – et grundstof velkendt fra behandling af stemningslidelser. I de seneste år har flere forskerteams observeret, at personer, der tager lave doser lithium, muligvis har en lavere risiko for at udvikle Alzheimers sygdom.
Tidligere arbejde viste, at lithium stabiliserer mikrotubuli – det vil sige, at det styrker “motorvejenes” struktur i neuronerne, selv når de omgivende forhold er ugunstige. Sammenholder man disse data med den nye teori, fremkommer en interessant konklusion: nøglen er måske ikke aggressiv fjernelse af plaques, men beskyttelse af selve cellens transportsystem.
Fremtidige terapeutiske strategier kan sigte mod at opretholde mikrotubulernes gennemgængelighed og genoprette balancen mellem beta-amyloid og tau, frem for udelukkende at fokusere på at nedbryde aflejringer. Forfatterne foreslår også at styrke autofagimekanismerne, så neuronerne bedre kan håndtere et overskud af “affaldsproteiner”.
Det kan betyde en helt ny generation af lægemidler – sådanne der regulerer cellens interne genbrugsprocesser frem for blot at fungere som “støvsugere” til amyloid. Blandt de lovende stoffer er rapamycin, metformin og resveratrol, som i prækliniske studier har vist evnen til at aktivere autofagi.
Andre kandidater er forbindelser, der stabiliserer mikrotubuli – på samme måde som taxol, der anvendes i onkologien. Forskellen ville ligge i dosering og specificitet. Målet er ikke at stoppe celledeling, men at understøtte stabiliteten af den neuronale infrastruktur. Forskere fra Massachusetts Institute of Technology og Johns Hopkins University tester allerede modificerede varianter af disse stoffer.
Hvad betyder den nye teori for fremtidige Alzheimer-patienter?
Hvis yderligere studier bekræfter denne model, vil læger muligvis begynde at betragte Alzheimer som en sygdom præget af dynamisk ubalance snarere end simpel ophobning. Diagnostikken vil i højere grad kunne tage hensyn til ikke blot mængden af plaques og klumper, men også tilstanden af mikrotubuli og autofagiens ydeevne.
Forestil dig to scenarier. I det første har en neuron allerede en betydelig mængde beta-amyloid, men dens genbrugssystem fungerer stadig, og mikrotubuli forbliver relativt stabile. Her kan en behandling, der styrker autofagien og stabiliserer mikrotubuli, holde cellen i live i lang tid. I det andet scenarie er autofagien praktisk talt kollapset, og beta-amyloid fortrænger massivt tau. I det tilfælde er selv meget effektiv “rensning” af plaques måske ikke nok, fordi selve neuronens interne infrastruktur allerede er ødelagt.
For personer i risikogruppen – eksempelvis med familiær disposition for demens – åbner denne tilgang nye muligheder for forebyggende indsats. En livsstil, der fremmer mitokondriernes sundhed, reducerer oxidativt stress og styrker cellernes generelle kondition, kan indirekte understøtte autofagien. Der pågår også studier af farmakologiske stoffer, der stimulerer cellulær genbrug og forbedrer mikrotubulernes stabilitet.
Mikrotubuli er en del af cytoskelettet – cellens indre konstruktion, der kan sammenlignes med et net af jernbaneskinner. Tau varetager sikkerheden på disse skinner. Beta-amyloid optræder i den diskuterede hypotese som en indtrænger, der forsøger at besætte de pladser, der er beregnet til tau. Lykkes det, bliver transporten af neurotransmittere usikker, og gradvist falder flere og flere transportlinjer ud af drift.
En sådan billedlig sammenligning hjælper med at forstå, hvorfor små molekylære forskydninger i proteinbalancen efter mange år kan føre til så dramatiske symptomer som hukommelsestab, desorientering eller personlighedsændringer. Alzheimers sygdom fremstår i lyset af den nye teori ikke som én enkelt katastrofe, men som en langvarig konflikt om hjernens nøgleinfrastruktur – en konflikt, der i årevis forbliver skjult, inden symptomerne bliver synlige udadtil.